I have ordered cookies to be delivered to our first ECL class. We will try to get to know each other and I will attempt to describe my research work and educational background for you, as well as what we should expect from this course. Then I will give you links to two surveys that will assess your programming knowledge and will determine the favorite programming language which the majority of class wish to learn.
About me, Amir, the instructor
I am a physicist and researcher, currently at Institute for Computational Engineering and Sciences at The University of Texas at Austin. I am a member of the Center for Computational Oncology led by Dr. Thomas E. Yankeelov and Dr. J. Tinsley Oden at ICES with the goal of understanding Tumor Growth.
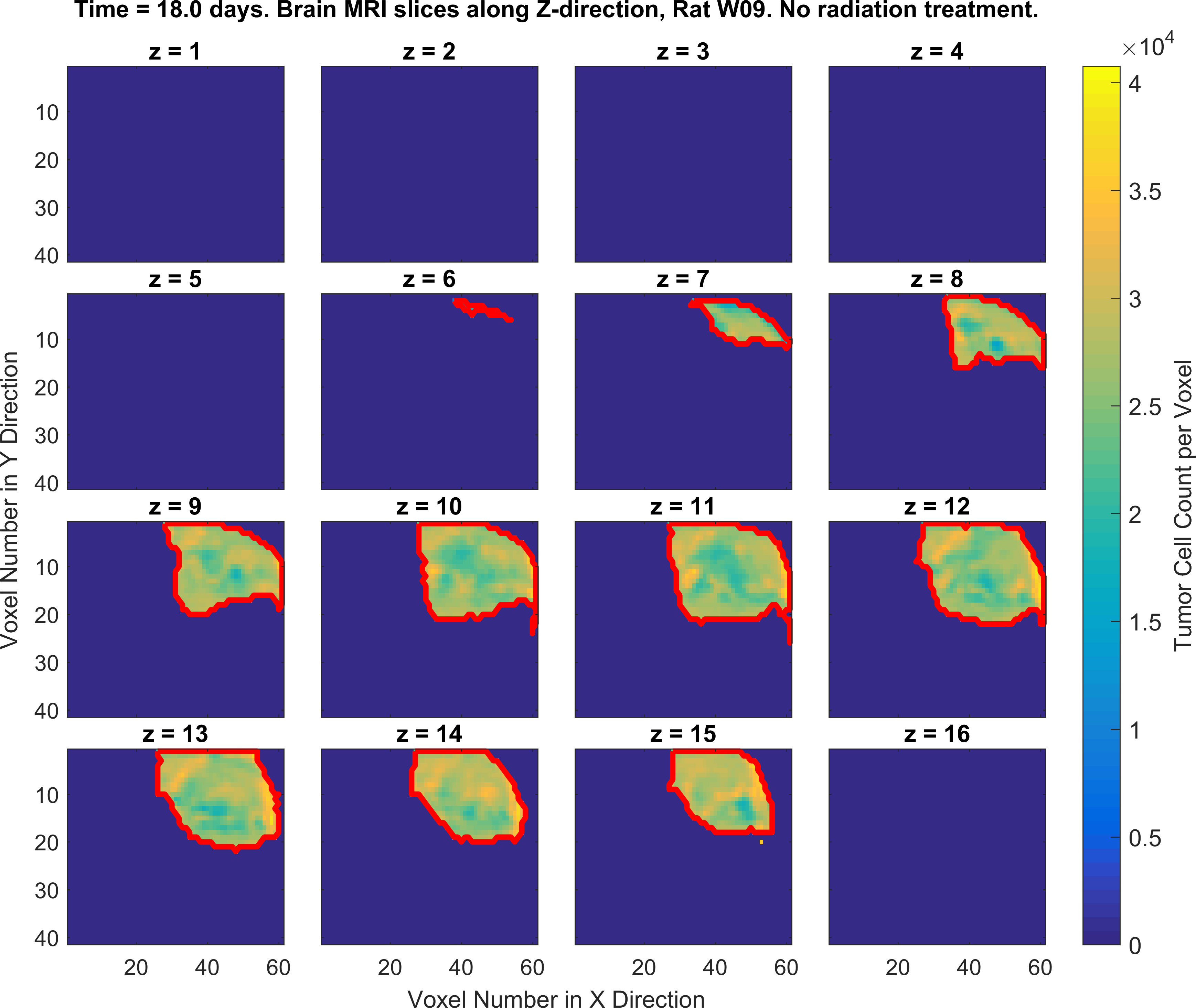
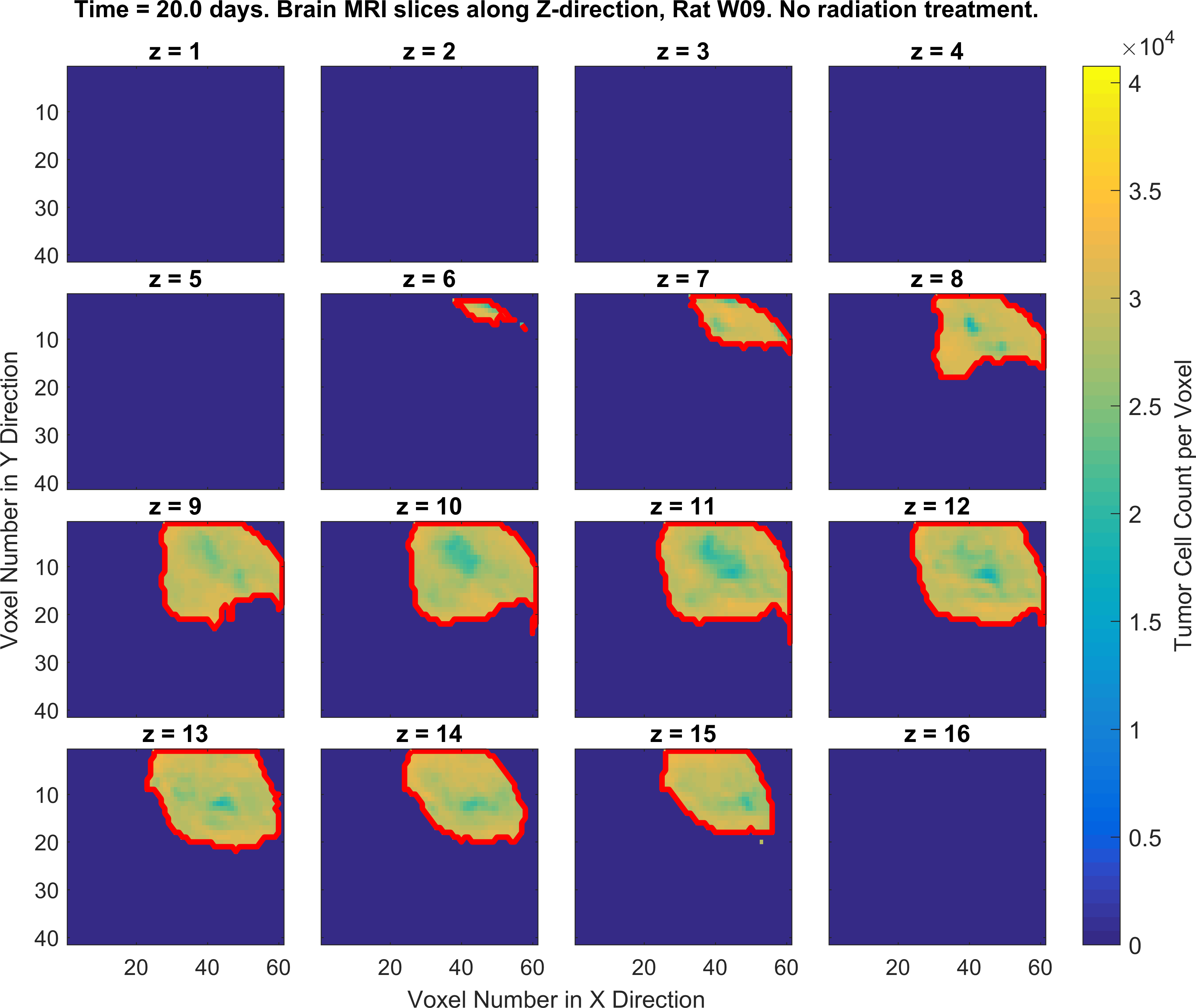
The following figures show the growth of Glioblastoma tumor cells in a Rat’s brain over time.
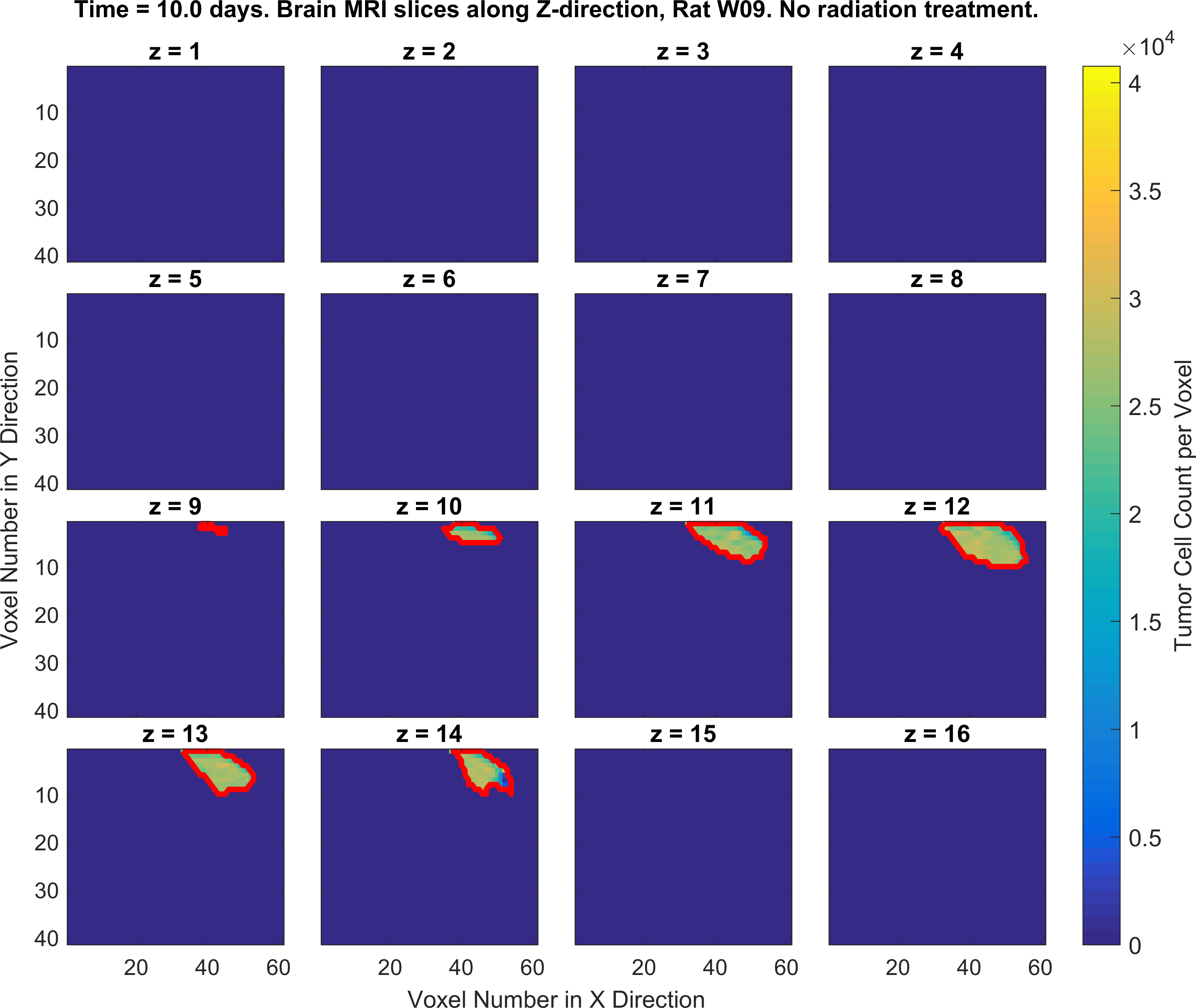
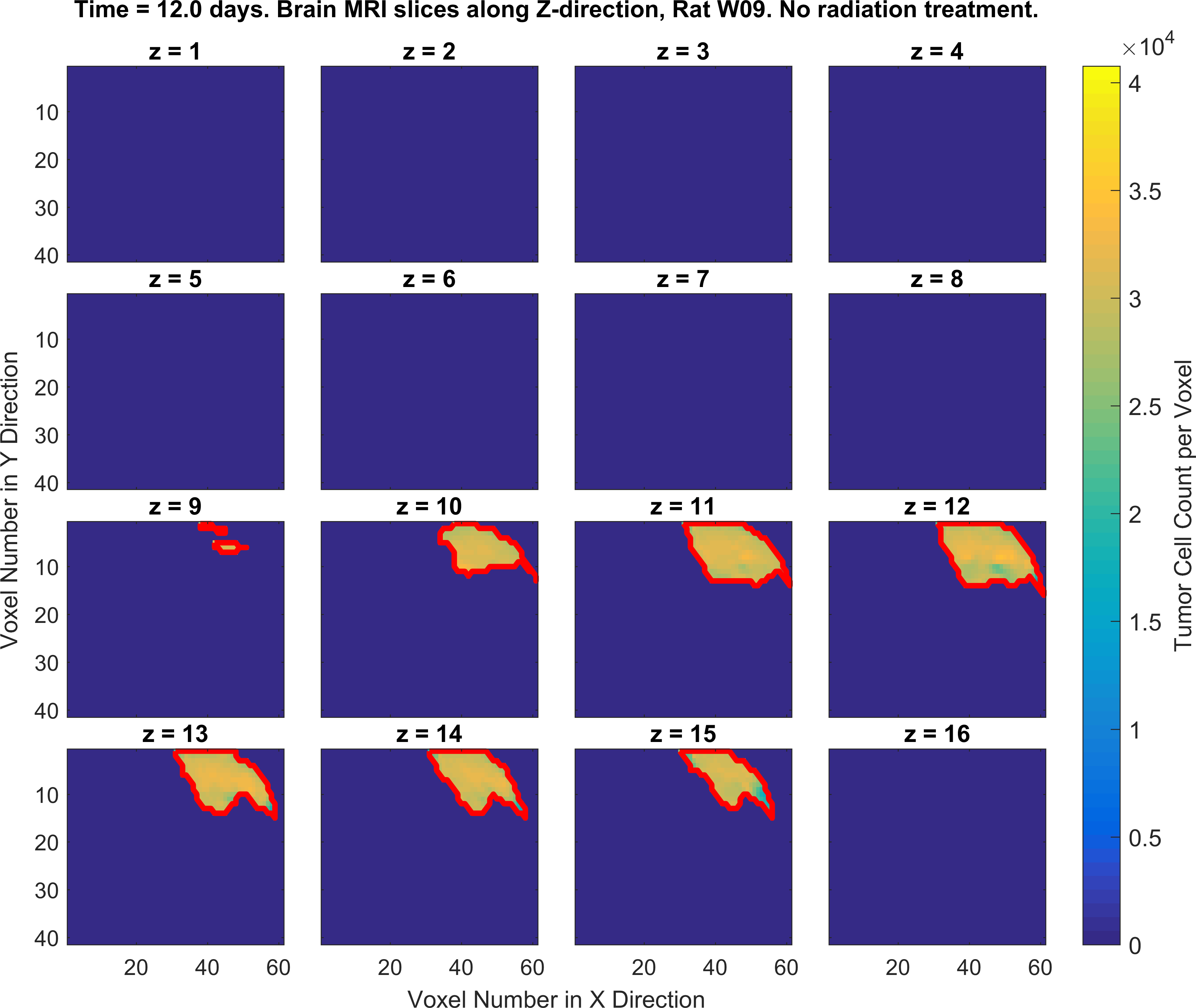
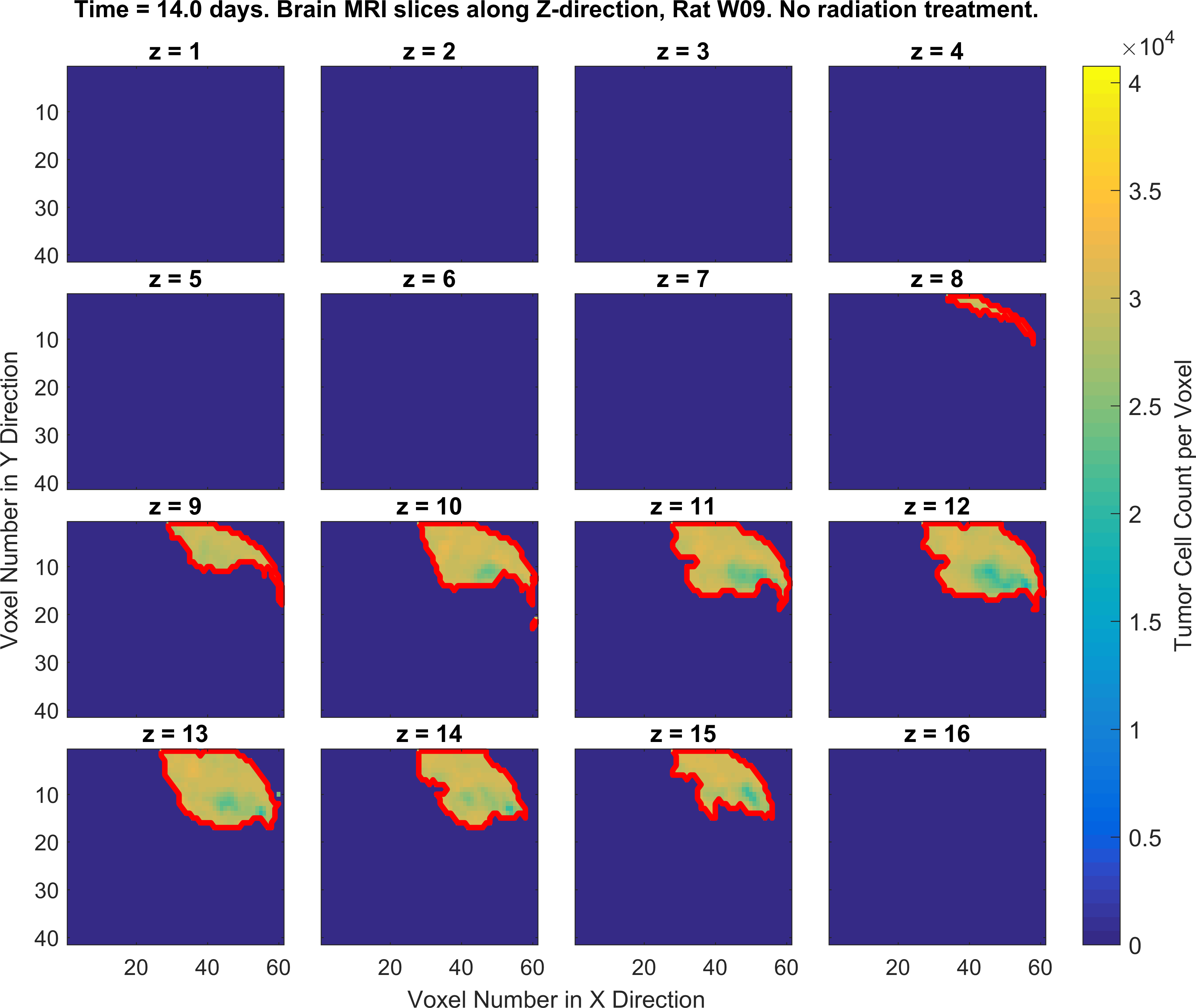
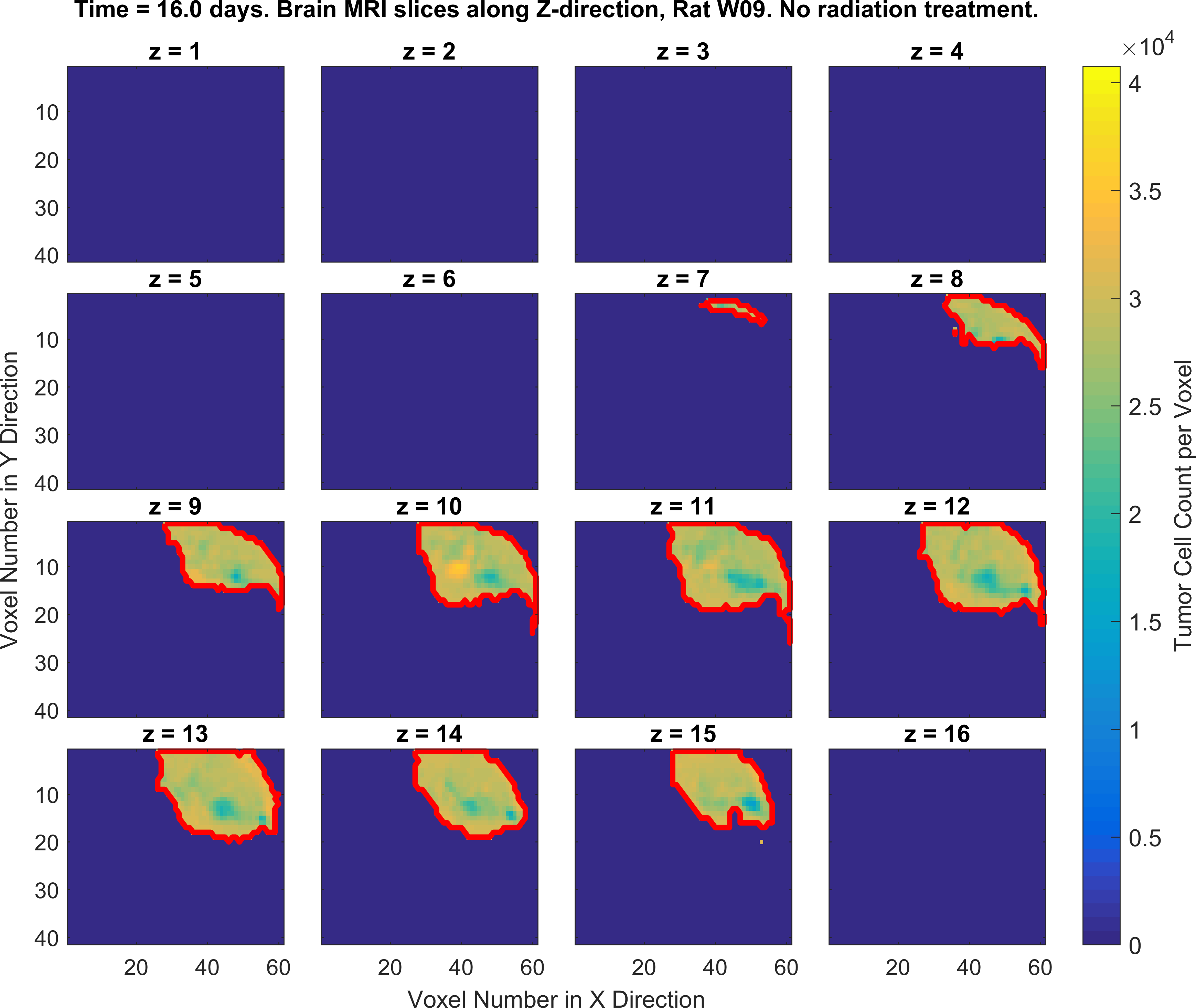
One of the fields on which my research is currently focused, is developing Monte Carlo optimizer/sampler and integrator algorithms for Bayesian inverse problems. Below you see example animations of two Markov Chain Monte Carlo (MCMC) samplers, both of which sample a double Gaussian-peak function, but with different MCMC sampling parameters.
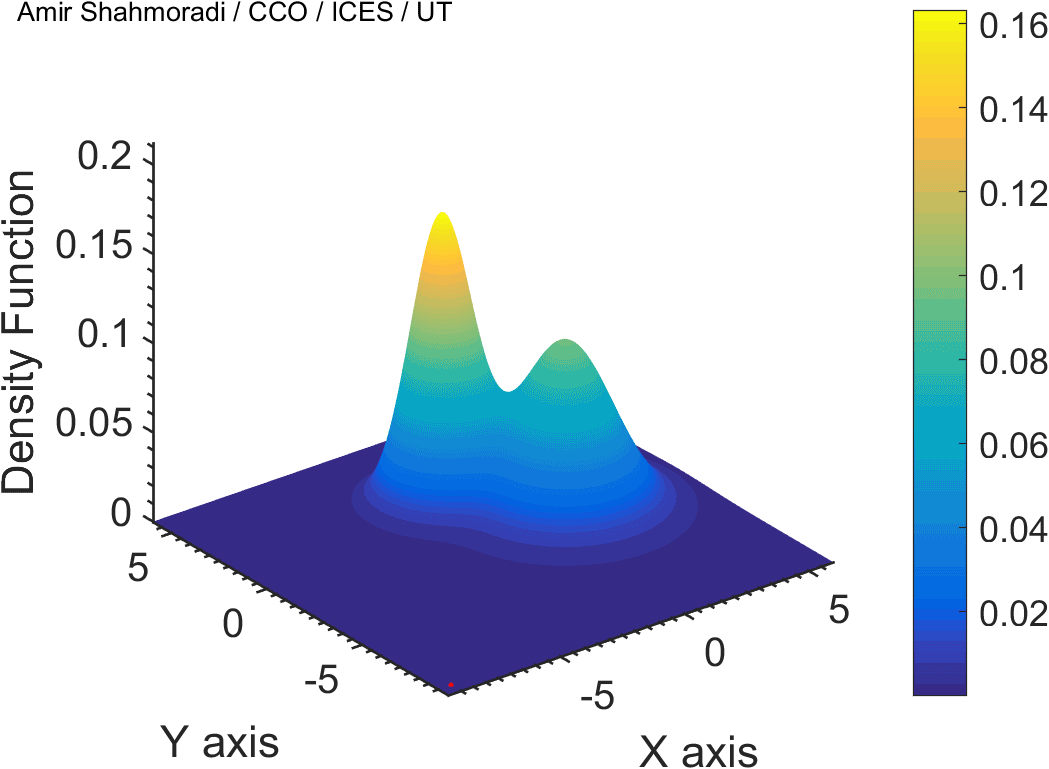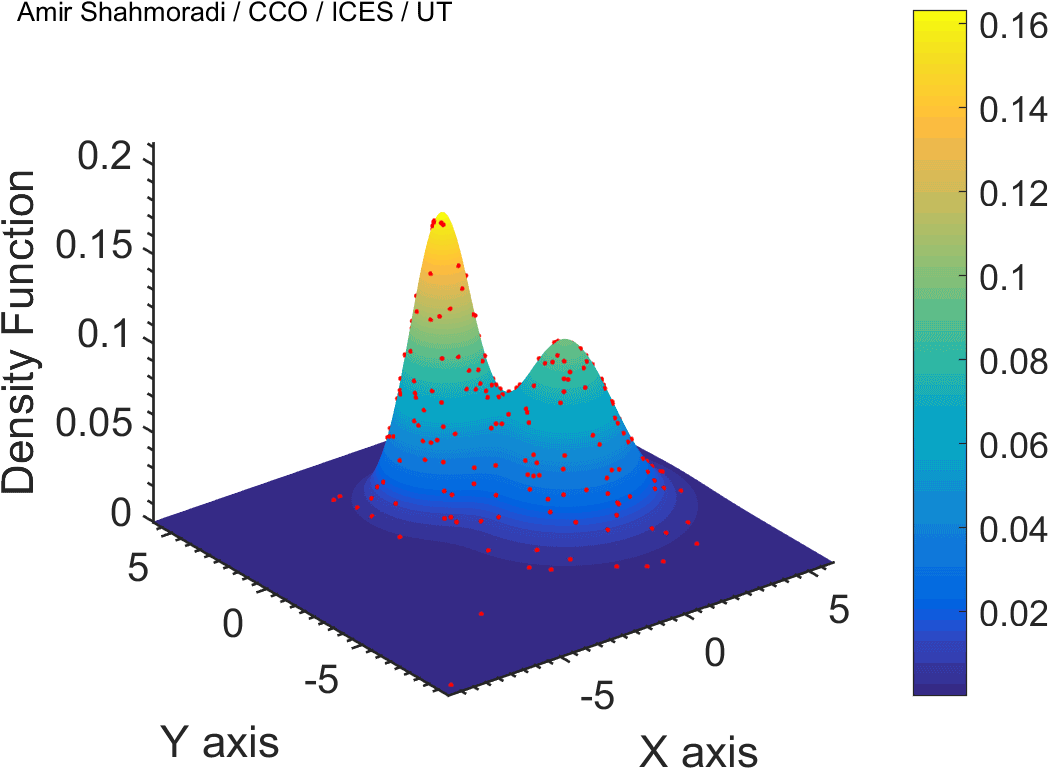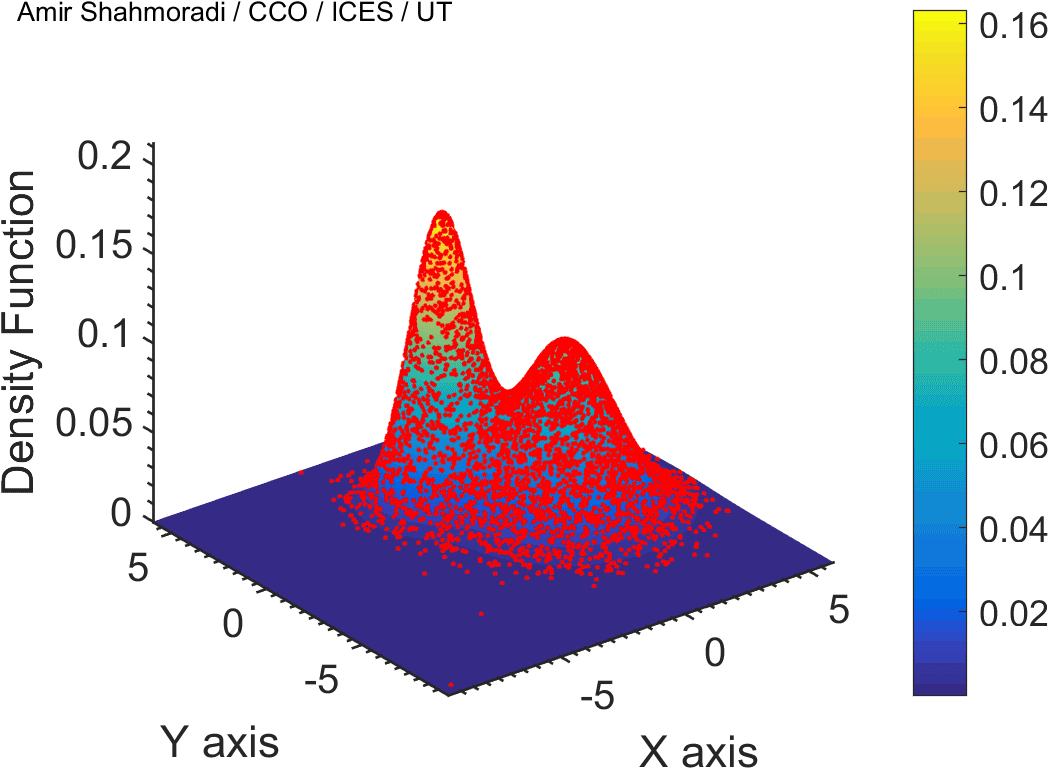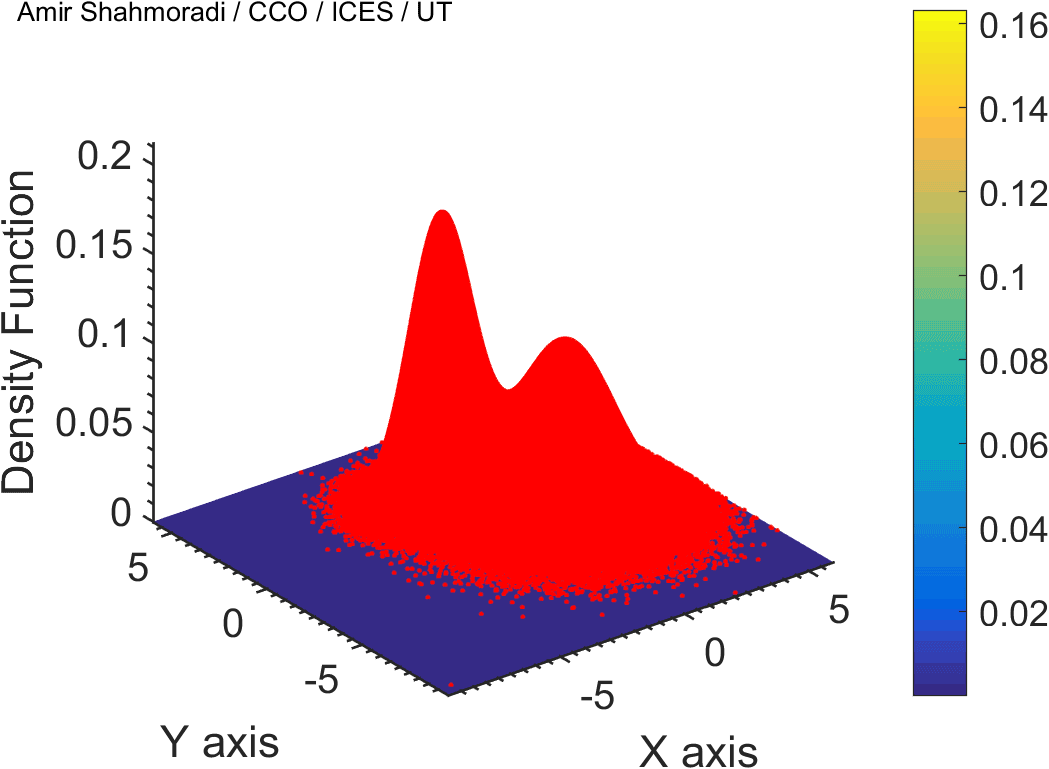
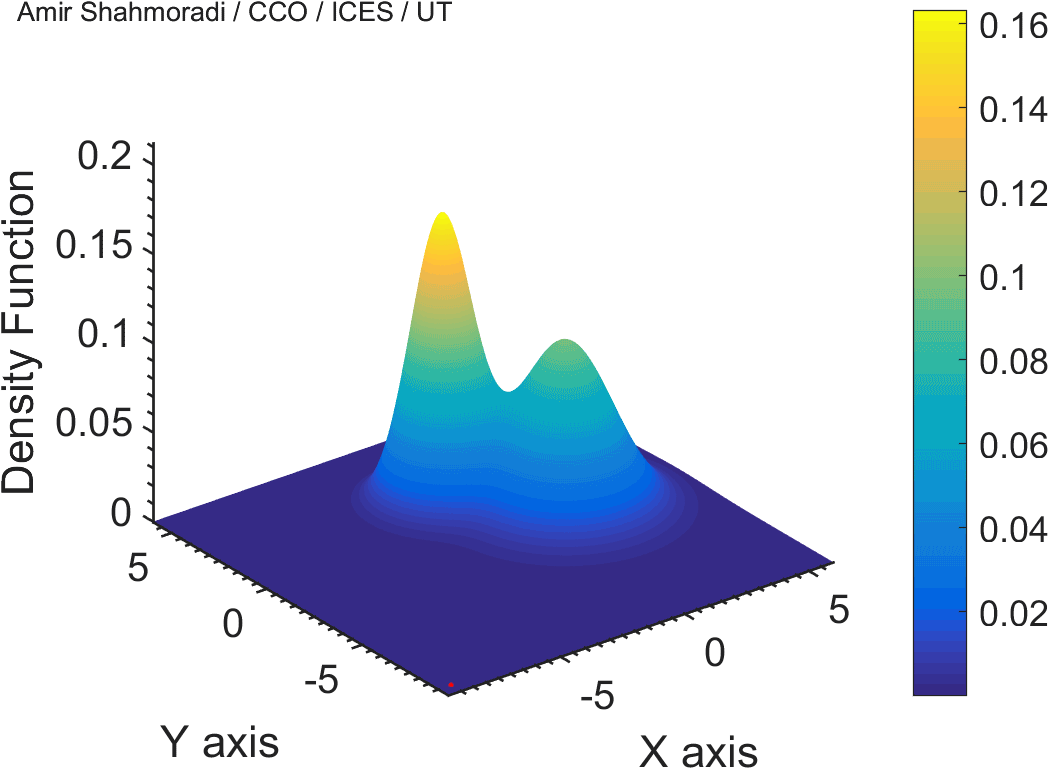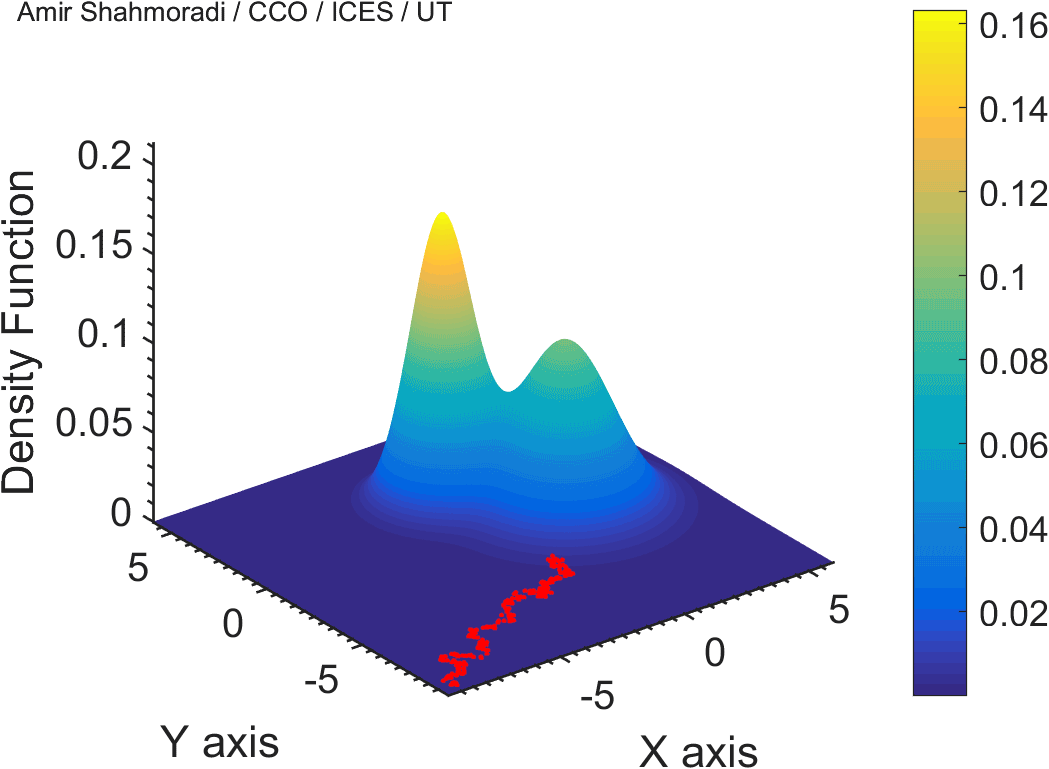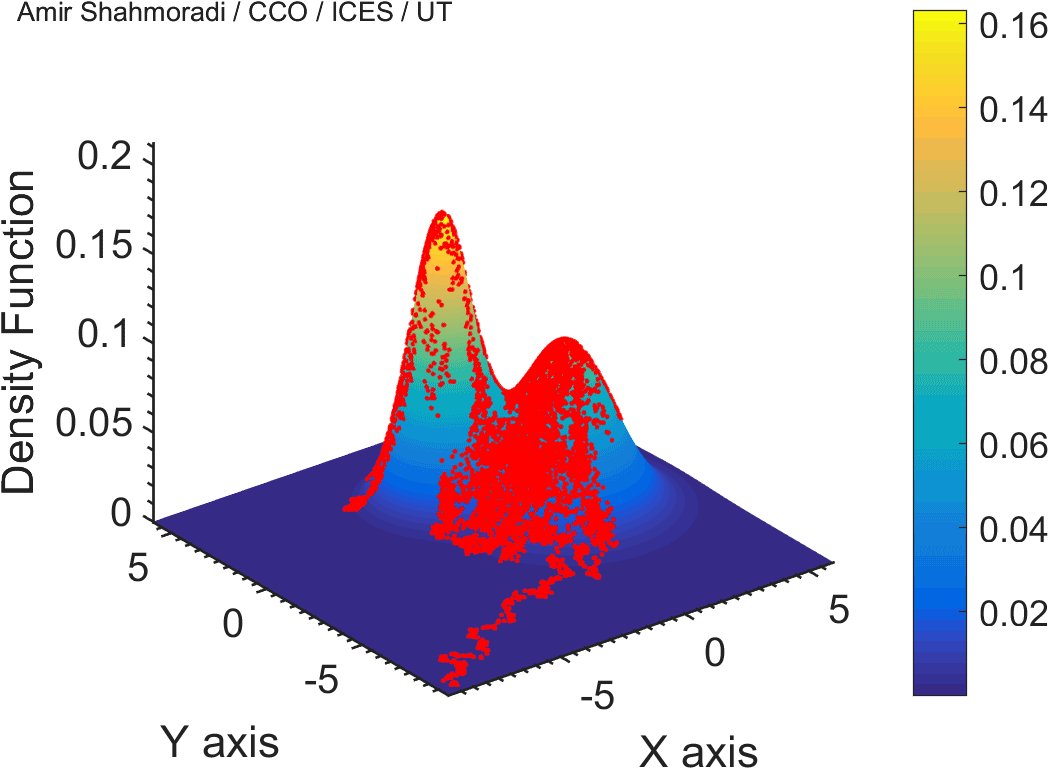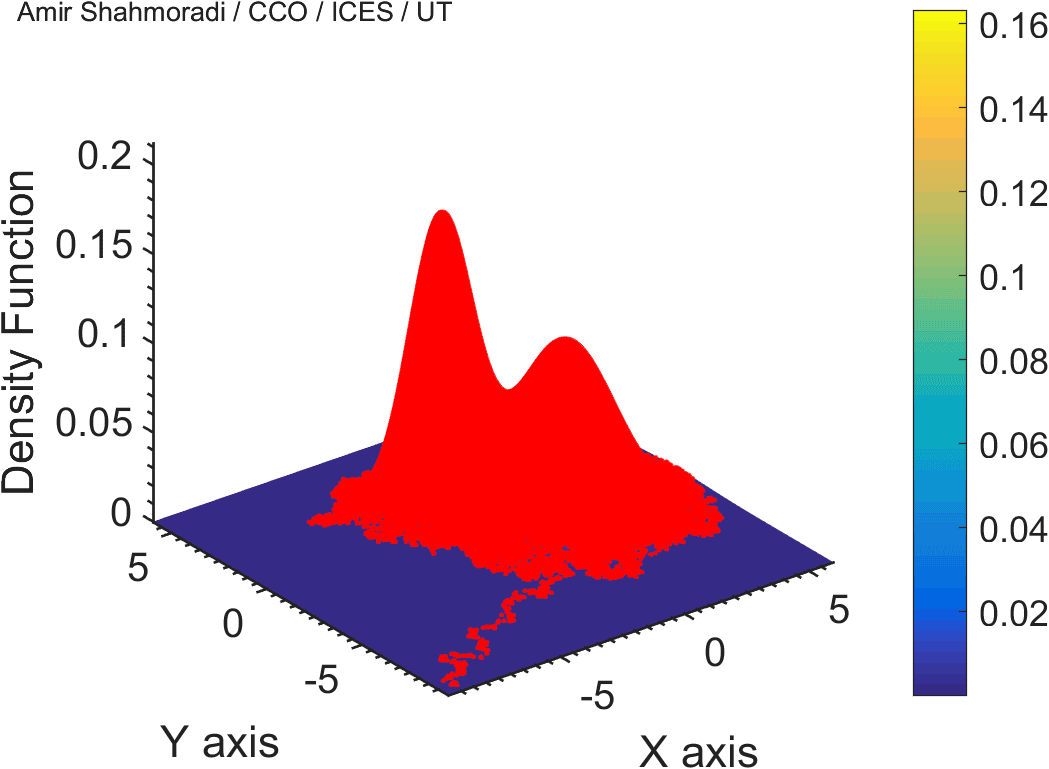
While my current focus of research is mathematical modelling of tumor growth and Monte Carlo samplers, I am and have been working in multiple branches of science and engineering for several years, from the subatomic world of elementary particles, to the microscopic world of biological macromolecules, to astrophysical phenomena occurring on the grandest scales of the observable Universe.
For several years of my research, I have been working on understanding Gamma-Ray Bursts (GRB) and their physics. Below is a movie of the moment a Short-duration GRB is generated from the merger of a binary Neutron star system.
I have also worked for a few years in the field of bioinformatics and evolutionary biology. The overarching goal in the field of protein bioinformatics and biophysics is to understand how proteins fold into their unique structure, and what determines the stability of the protein 3D structure. One of the workhorses of this field, is therefore molecular dynamic simulation to probe the dynamics of proteins and their interactions with other molecules. The following is a 1.5ns molecular dynamics simulation of Human Influenza H1 Hemagglutinin protein (1RD8, chains AB).
